Our research interests are centred around materials used for renewable energy generation (e.g. solar cells) and storage (e.g. reusable batteries). We use a branch of physics called Density Functional Theory (DFT) to predict the properties of these materials and link the macroscopic observables (such as open circuit voltage or thermodynamic stability) with microscopic processes (such as electron capture or electron-phonon coupling).
DFT is an ab-initio (first-principles) method derived from quantum mechanics and can be used to predict material properties without experimental input (see, for example, this open access article). Our atomic scale models can be used to rationalise existing experimental observations, or guide future investigations. For example, it can explain why heat travels slowly through some materials or predict new materials for high-performance solar cells.
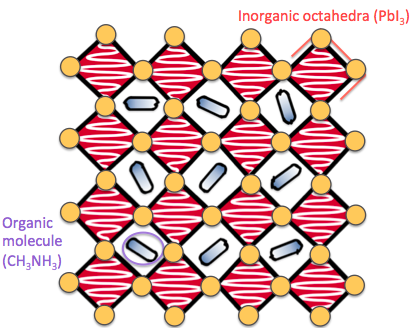
When DFT is applied to crystalline materials it is usually assumed that there is perfect translational symmetry - that there are no defects (missing or extra atoms) - and that the atoms are perfectly static. However a material always has defects (these are unavoidable due to the laws of thermodynamics [1]), and the atomic lattice vibrates with heat. These defects and vibrations are important to understand because they can have a significant impact upon the performance of a device. Our research has focused on the defects and lattice distortions in halide and chalcogenide perovskite materials, a family of materials that have become incredibly popular over the last decade as they can convert sunlight into electricity efficiently, and have the potential to form more flexible, lightweight and cheaper solar panels than those currently on the market.
[1] This is because the cost in lattice energy is balanced by the change in entropic energy.